靶向FGFR4的抗肿瘤药物研究进展
发表时间:2017-04-13 20:49:00
39%的食管鳞状细胞癌患者和82%的恶性外周神经鞘膜瘤患者存在FGFR4异常过表达,伴随着不良预后和短的生存期。
成纤维细胞生长因子受体4(FGFR4)通过启动STAT3、MAPK和PI3K/AKT等信号级联,调节细胞的增殖、分化和转移。其基因突变、过表达或其配体分子过表达等异常信号转导在某些肿瘤的发生发展中发挥着重要的作用,表明FGFR4是此类肿瘤潜在的治疗靶标。靶向FGFR4的抗肿瘤药物或方法主要包括小分子抑制剂、单克隆抗体、配体捕获蛋白、短链RNA寡核苷酸适配体(shRNA)。本文回顾FGFR4异常的肿瘤及靶向FGFR4抗肿瘤药物的研究进展。
FGFR(fibroblastgrowthfactorreceptor)酪氨酸激酶家族包括FGFR1、FGFR2、FGFR3和FGFR4。其由胞外变异区、结合硫酸乙酰肝素蛋白聚糖的保守区、FGF结合区、单次跨膜区及胞内酪氨酸激酶区组成。大部分FGFs在共同受体Klotho的帮助下与FGFRs和肝素形成复合物,导致构象改变的FGFR胞内激酶区自磷酸化而活化STAT3信号通路。自磷酸化的FGFR也可以磷酸化其适配体蛋白FRS2α,活化Grb2/Sos1复合物而启动下游MAPK和PI3K/AKT信号通路。此外,FGFR以FRS2α非依赖的方式活化磷脂酶C-γ(PLC-γ),磷酸化RAF,加强MAPK信号,发挥其调节细胞增殖、分化和转移的功能。MAPK信号通路主要跟FGFR介导的细胞增殖和转移相关,而PI3K/AKT信号通路主要跟细胞的运动性和存活相关。在生理状态下,FGFR4信号通路被严格控制,而FGFR4信号失调导致癌症的发生发展、增殖、存活及转移。本文回顾FGFR4异常的肿瘤及靶向FGFR4抗肿瘤药物的研究进展。
1FGFR4异常的肿瘤
1.1基因突变
FGFR4激酶域激活突变在横纹肌肉瘤、恶性肺腺癌、恶性胶质瘤等肿瘤中均有发现。7%~8%横纹肌肉瘤患者中存在FGFR4激酶活性持续激活的突变,其突变位点主要发生在K535和E550,增加了FGFR4激酶活性,通过STAT3途径活化下游信号通路,但其磷酸化Akt和ERK能力的下降,往往导致具有侵袭性的晚期肿瘤。然而,肿瘤细胞中FGFR4的突变位点不仅仅发生在激酶域。研究表明,乳腺癌细胞MDA-MB453的FGFR4Y367C突变引起FGFR4自发形成二聚体,从而导致组成型活化ERK,增强MAPK信号通路,促进细胞增殖,诱发肿瘤的形成,且对其配体和抗体的刺激均无反应。此外,含FGFR4等位基因Arg388或纯合子的肿瘤细胞通过使FGFR4受体内化,导致FGFR4异常信号持续激活,增加乳腺癌的侵袭,降低鳞状细胞癌、肺癌患者存活率,增加前列腺癌恶性程度,增加黑色素瘤的厚度。
1.2过表达
39%的食管鳞状细胞癌患者和82%的恶性外周神经鞘膜瘤患者存在FGFR4异常过表达,伴随着不良预后和短的生存期。在乳腺癌患者中大约有32%FGFR4的mRNA表达水平上升,其中有10%是由于FGFR4基因扩增所导致的过表达,且与雌激素受体和孕激素受体水平及淋巴结转移正相关。而在星形胶质细胞瘤中,FGFR4过表达预示着癌症晚期和短的生存期。与良性前列腺增生相比,前列腺癌患者FGFR4的表达水平显著上升,且其表达水平与格里森评分正相关,并伴随着不良转归。大约三分之一的肝癌患者存在FGFR4异常高表达且预后差。此外,FGFR4过表达的卵巢癌、胃癌、胰腺癌、结肠癌有较高的侵袭性,而抑制FGFR4信号通路能显著降低其侵袭性,表明FGFR4是潜在的治疗靶标。
2药物研发进展
通过阻断胞外配体分子与受体的结合或胞内激酶信号的传递,抑制FGFR4所介导的增殖信号,是治疗此类肿瘤的有效手段。其方法包括小分子激酶抑制剂、单克隆抗体、配体捕获蛋白和shRNA等。近年研发的靶向FGFR4的抗肿瘤药物及其适应证见表1。
小分子抑制剂
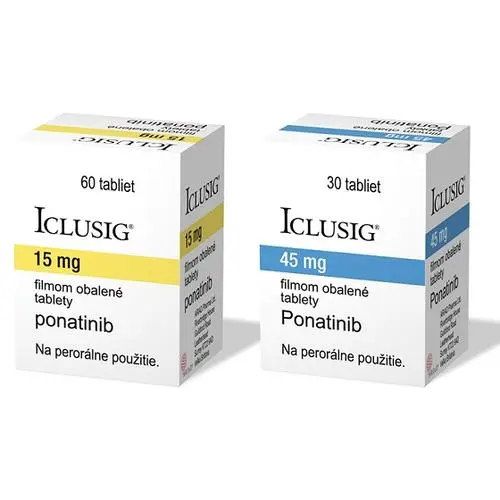
小分子酪氨酸激酶抑制剂通过阻断胞内激酶与ATP结合的活性,阻断细胞增殖信号。FGFR4的小分子抑制剂可分为泛FGFR和FGFR4特异性小分子抑制剂。由于FGFR1、FGFR2、FGFR3激酶域的结构相似,现阶段研发的针对这三个激酶的抑制剂效果相差不大。然而,FGFR4激酶域与FGFR1-3激酶域存在一定差异,因此许多能有效抑制FGFR1-3的抑制剂对FGFR4效果不佳。如CH5183284、BGJ398和AZD4547等进入临床Ⅰ期或者Ⅱ期小分子抑制剂对FGFR1-3的选择性(IC50<10nmol/L)远高于FGFR4。而JNJ-42756493和LY2874455是少数对FGFR1-4具有同等高效抑制效果的泛FGFR小分子抑制剂,其IC50均达到个位数纳摩尔级别。JNJ-42756493和LY2874455在细胞中均通过抑制FGF/FGFR信号通路,表现出FGFR依赖性抗增殖作用,对FGFR异常的移植瘤具有极强的抑制作用,且呈剂量效应抑制肿瘤生长。JNJ-42756493的临床Ⅰ期试验(NCT01962532)结果确定其指导临床Ⅱ期的用药剂量(RP2D)为10毫克/日(用药7天,停药7天)。LY2874455的临床Ⅰ期试验(NCT01212107)确定其RP2D为16毫克/日,1日2次。在AZD4547的临床Ⅰ期试验中(NCT00979134),AZD4547对FGFR基因扩增的鳞状非小细胞肺癌患者显示强肿瘤杀伤活性,且在80mg的剂量下有很好耐受性。而Ponatinib治疗FGFR异常的晚期肺鳞癌患者的临床Ⅱ/Ⅲ期试验(NCT01761747)因不良反应而终止。
缺乏选择性的FGFR激酶抑制剂因脱靶而导致高磷酸盐血症、指甲脱离、脱发、黏膜炎、味觉障碍和黏膜干燥、结膜炎、角膜炎、眼睛干燥、无症状视网膜色素层剥离、骨关节疼痛、肌痛等不良反应,限制其临床应用。为了提高小分子抑制剂对FGFR4激酶域的选择性并减少不良反应,阿斯利康研发了FGFR4选择性抑制剂AZ709。其体外实验对高水平表达FGF19或FGFR4的细胞显示出很好抑制效果,但体内实验无明显作用。诺华公司的FGFR4选择性抑制剂FGF401能特异性靶向FGFR4,治疗其过表达的肝癌等恶性肿瘤,目前正在临床Ⅰ/Ⅱ期试验的患者招募阶段(NCT02325739)。H3生物医药公司的FGFR4特异性抑制剂H3B6527对FGF19基因扩增的细胞具有强抗肿瘤活性,且在小鼠和猴的动物模型中没有胆汁酸相关的不良反应。最近,蓝图医药[11]研发并报道了一个全新的、高选择性的、不可逆结合的FGFR4小分子抑制剂BLU9931,IC50为3nmol/L。其选择性比作用于FGFR1/2/3分别高297、184和50倍。FGFR4/BLU9931复合物晶体结构显示BLU9931与FGFR4铰链区C552形成共价键,苯胺喹唑啉基团与A553形成两对氢键。二氯二甲氧基苯基团选择性占据FGFR4疏水口袋,苯胺的苯环形成一个大约60°的二面角,使邻位的丙烯酰胺正对着C552的巯基,而这个位置与FGFR1-3相同位置的酪氨酸形成了空间位阻。BLU9931有效抑制FGFR4信号通路的磷酸化,抑制肝癌细胞系的增殖。在Hep3B细胞的小鼠移植瘤模型中,口服BLU9931成剂量关系抑制肿瘤的生长,其中有2只小鼠(共9只)治疗30天后肿瘤消失。相比于BLU9931,临床上治疗肝癌的标准药物索拉菲尼的抑制效果较差,且导致小鼠体重下降。而BLU9931治疗组未出现小鼠体重下降的不良反应。此外,蓝图医药的另一个FGFR4特异性的抑制剂BLU554也进入临床Ⅰ期试验(NCT02508467),治疗FGFR4过表达的肝癌和胆管癌,正处于患者招募阶段。